Lipoprotein Tutorial
Click on 'Model' in the menu to the left to view this at any time. You can select and print a copy from this page. The model depicts the interrelationships between the major topics of this tutorial.
Click on "Overview" shown in the Menu on the left.
Disclaimer
The physiology of lipoproteins has been actively researched for the past few decades. Research has answered many questions about how this system works but even more questions have evolved as a result. Lipoprotein physiology is still a hot topic today.
Accordingly it has been very challenging to construct a diagram that shows the interactions among components. The information presented here is meant to give the general student a basic understanding of lipoprotein interactions and is not intended to show all that is understood about this complex system. Indeed, all is not understood.
September 2013
Lipoprotein Overview
The most obvious reason that too much cholesterol is 'bad' is that it contributes to the formation of atherosclerotic plaques. As these enlarge they constrict the artery and impeed blood flow. If a plaque ruptures small pieces of it (thrombi) enter the circulation and become lodged in small downstream vessels. If such a vessel is in the brain the result is a stroke; if in the heart the result is a heart attack.
Not so obvious is the effect excess cholesterol has on all body cells. Though cholesterol is a normal component of cell membranes and contributes to their stability, excess cholesterol causes rigidity. This impedes the normal and necessary lateral movements of membrane components affecting activities such as enzyme function, endocytosis, cell growth, etc.
Cholesterol
Body cells can synthesize enough of their own cholesterol to keep pace with everyday demands. If they need more they position LDL receptors in their membranes and engulf cholesterol-rich LDLs to fulfill this need. If they have excess cholesterol they utilize SR-B1 and ABCA-1 transporters to move cholesterol to the outer leaflet of the cell membrane. From this locations HDL particles pick up cholesterol for transport to the liver where it will be secreted as bile.
Triglycerides
In addition to being synthesized by body cells, dietary cholesterol is brought into the body as a minor component of chylomicrons; the major component is triglyceride. These largest of all lipoproteins deposit triglyceride in different regions of the body depending on when the individual has last eaten. Just after a meal triglycerides will be hydrolyzed into fatty acids and monoglycerides in adipose tissue for storage. When fasting this will occur in muscle tissue to be used for energy.
The now smaller chylomicrons called remnants will be taken into the liver and removed from circulation. The liver will recirculate most triglycerides and a small amount of cholesterol in VLDLs. This will maintain a supply of triglycerides for skeletal and heart muscle between meals. VLDL remnants bypassing the liver interact with other circulating lipoproteins and exchange triglycerides and cholesterol.
Low Density Lipoproteins
Why is cholesterol circulating around the body in chylomicrons and VLDs if it isn't deposited somewhere? And anyway, the smallest lipoproteins -- the remnants -- can't penetrate capillary walls to reach any body cell that does have LDL receptors displayed.
With the help of hepatic lipase the IDL (VLDL remnant) can lose its remaining triglyceride and become small enough to penetrate capillary walls. This is particularly important to body cells that require a good supply of cholesterol to synthesize steroids (testosterone, cortisone, aldosterone) However, hepatic lipase is bound to hepatocyte membranes in an inactive form and must be released, circulated and activated in order to convert IDLs to LDLs.
HDL2 particles, especially those that have picked up some triglyceride from lipoprotein-rich particles, are responsible for displacing inactive hepatic lipase from hepatocytes. They then transport the still inactive enzyme through the body until contacting triglyceride-rich lipoproteins. Hepatic lipase is transferred to these particles and thus becomes activated. This enzyme hydrolyzes any triglycerides in the particle and also breaks down excess phospholipids in the coat. If the triglyceride-rich lipoprotein is an intermediate density lipoprotein (IDL) the resulting smaller particle could be an LDL. LDLs are able to penetrate capillary walls.
They are also small enough to slip beneath the lining of arteries that have become become inflamed. Macrophages in these areas release chemicals that alter the LDLs causing them to become oxidized. The macrophages also have special SR-A receptors that specifically engulf oxidized LDLs. This cholesterol uptake by the macrophages converts them into foam cells that mark the early stages of atherosclerosis.
High Density Lipoproteins
Now that the problem of delivering cholesterol to body cells -- and inflamed arteries -- has been solved the new problem is how to retrieve excess cholesterol. This is where HDL comes into play once more. This lipoprotein is small enough to pass through all capillary walls and also to reach foam cells. In addition to SR-B1 transporters, foam cells display ABCG-1 transporters both of which move intracellular excess cholesterol to the surface. Because HDLs don't have much free cholesterol in their coat there is a cholesterol gradient between their coat and cells displaying excess cholesterol. When in contact, this gradient facilitates cholesterol movement to HDLs. The HDLs return the cholesterol to the liver (reverse cholesterol transport) for secretion as bile.
As you read through each section below, scroll back up to the top of the page
and locate the part
of the model being described.
Design of the Model
The large uncolored at circle center left is the liver.
Lavender means triglycerides & orange means cholesterol esters.
Large colored circle at center top is a chylomicron or a VLDL.
Two smaller two-colored circles beneath it are remnants.
Muscle, adipose, body cells, liver & intestines are labeled; foam cell represents arthrosclerosis.
Dark blue labels are membrane bound proteins.
Red labels are plasma enzymes or transporters.
Small colored circles with black labels are various lipoproteins.
Lipoproteins come in a range of sizes and contain triglycerides and cholesterol esters. They are classified according to their relative amounts of heavy protein and light lipid.
Structure & Density Classification
Lipoprotein Structure
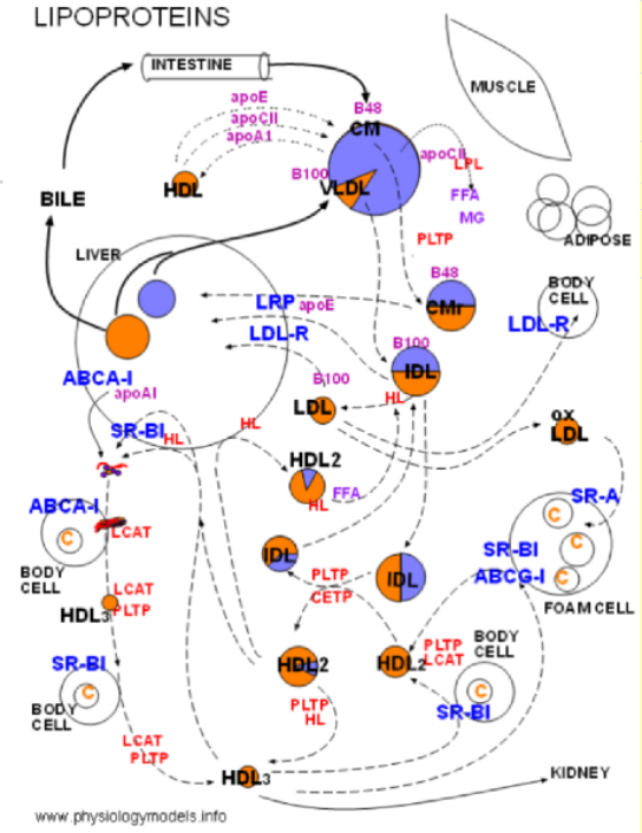
The left illustration shows that lipoproteins consist of a core containing triglycerides (lavender) and cholesterol esters (orange). This is surrounded by a single-layered coat of phospholipids (purple) and free cholesterol (orange) shown in the expanded segment. There are also proteins (red) attached to the coat; these are called apoproteins.
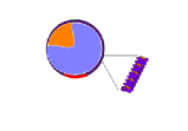
The illustration to the right shows lipoproteins of various sizes going from large, triglyceride-rich ones to small cholesterol ester-rich ones. The relative amount of triglyceride (lavender) and cholesterol ester (orange) is shown. The red particle on the surface of each represents an apoprotein
The largest lipoprotein is the chylomicron (1) from the intestine. The liver also secrets a relatively large one called a very low density lipoprotein (VLDL) represented by 4. Lipoproteins 6 and 7 represent high density lipoproteins (HDLs); they are cholesterol-ester rich as indicated by their mostly orange core.
VLDLs and chylomicrons lose their triglycerides (not their cholesterol esters) as they circulate though the body. Illustration 2 and 3 are representative of a chylomicron that is progressively losing triglyceride. It will continue to shrink until resembling 5 known as a chylomicron remnant. VLDLs (4) also lose triglycerides until they become similar to 5 but these are called intermediate density lipoproteins (IDLs) -- both have about equal amounts of triglyceride and cholesterol ester in their cores.
The two particles at the far right represent high density lipoproteins (HDLs). Their main component is cholesterol ester. Unlike the triglyceride-rich lipoproteins described above, they start out very small and progressively get larger. The ones shown here represent the larger ones -- small ones are too small to show.
Density Classification
Lipoproteins are classified according to their density or 'weight per unit volume'. The apoprotein molecules are much heavier than any other type of molecule involved. As the lipoprotein shrinks the heavy apoproteins occupy relatively more of the particle's volume. Scan the lipoproteins 1-7 and notice the constant size of the apoprotein (red). Large lipoproteins have relatively less apoprotein than small ones so they have low density; smaller ones have higher density.
When referring to a specific lipoprotein as having 'low density' it is important to specify 'relative to which other lipoprotein'. In the illustration it would be correct to say that particle 3 has low density -- compared to particle 5 -- but, it has high density compared to particle 1. Terms such as HDL (high density lipoprotein), IDL (intermediate density lipoprotein), LDL (low density lipoprotein) and VLDL (very low density lipoprotein) are general terms and don't refer to a particular particle. Rather, they refer to subgroups of lipoprotein particles of similar densities.
Small Dense vs. Fluffy LDLs
If the smaller sized particles are abundant the likelihood of developing coronary artery disease is increased. This is often referred to as pattern B; pattern A has relatively fewer small LDLs. The smaller particles are referred to as small dense LDL (sdLDL) while particles on the larger end of the scale are referred to as 'fluffy'. The clinical test that measures this distribution is called LDL-P ... 'p' stands for particles.
The largest circle in the diagram represents the liver. The small lavender circle within it represents stored triglycerides and the orange circle represents stored cholesterol (cholesterol esters). The mid-sized circle that has a triglyceride-rich (lavender) core represents the large lipoproteins that come from the intestines (chylomicrons) and from the liver (very low density lipoproteins, VLDL). The chylomicron has a specific apoprotein (B48) tightly bound to its coat; likewise the VLDL also has a similar bound apoprotein (B100). Both types of lipoprotein go through the same sequence of events -- hydrolysis of their triglycerides -- so the same particle represents them both. However, recall the size comparisons presented in the previous tutorial -- a chylomicron is significantly larger than a VLDL.
Lipoprotein Lipase
Lipoprotein lipase (LPL, red) is synthesized primarily by skeletal muscle and adipose tissue.
It is secreted into the interstitial fluid then diffuses to nearby capillaries. It becomes
attached to large, branched proteins (heparin sulfate
proteoglycans (HSPGs) bound to the inner capillary surface (not shown).
 Its function is to hydrolyze triglycerides (lavender) from lipoproteins into free fatty acids (FFA) and
monoglycerides (MG).
Its function is to hydrolyze triglycerides (lavender) from lipoproteins into free fatty acids (FFA) and
monoglycerides (MG).
However, lipoprotein lipase is inactive until it becomes bound to its cofactor, apoprotein CII (apoCII). Apoprotein CII is attached to VLDLs when they are secreted from the liver. Chylomicrons(CM) don't initially have this apoprotein but receive it (and apo E) from circulating high density lipoproteins (HDL)(see small-dashed arrows upper left). In exchange, the chylomicron transfers its apoprotein A1 to the HDL.
When the apo C-II on a triglyceride-rich lipoprotein contacts LPL on the capillary inner surface (note diagram) the lipolipase becomes activated and it hydrolyzes (dotted arrow) the triglyceride in the lipoprotein into free fatty acids(FFA) and monoglycerides (MG). The lipoprotein and attached enzyme can detach from the capillary wall and circulate; the hydrolysis continues as long as the enzyme remains attached.
The dashed arrows pointing from the triglyceride-rich lipoprotein to the two smaller circles indicates that lipoproteins shrink as the lose their triglycerides. As this occurs excess coat material (cholesterol, phospholipids, apoproteins) is picked up by phospholipid transfer protein (PLTP). With the loss of apo C-II, during the decrease in size, hydrolysis will cease.
The smaller lipoproteins are called 'remnants'; the remnant of VLDL is also called intermediate density lipoprotein (IDL). Their cores have approximately equal amounts of cholesterol ester and triglyceride (note colors in diagram.)
Regulation
The concentration of lipoprotein lipase in the above-mentioned tissues varies with the nutritional state at the time. This is best understood by considering blood glucose levels. After a meal (postprandial) a high glucose level stimulates the pancreas to release insulin into the blood. In addition to facilitating glucose uptake by cells, insulin also affects triglyceride metabolism.
The effect of insulin on adipocytes is to stimulate them to produce and secrete lipoprotein lipase. This facilitates the uptake of free fatty acids and monoglycerides into this tissue for conversion back to triglycerides for storage. Simultaneously, the effect of insulin on skeletal muscle is to inhibit the secretion of lipoprotein lipase. Overall this encourages triglyceride storage in adipose tissue rather than oxidation in muscles.
When fasting (low blood glucose), glucagon instead of insulin is the major hormone involved. The effect of glucagon on skeletal muscle is to stimulate the production and secretion of lipoprotein lipase This encourages free fatty acids to be oxidized for energy. Glucagon has no effect on adipose tissue.
Under stressful conditions, when adrenalin is circulating, both skeletal and cardiac muscles are stimulated to produce and secrete lipoprotein lipase. This supplements the glucose oxidation normally occurring under these circumstances.
VLDLs and chylomicrons shrink as they lose triglyceride and become 'remnants'. These become bound to receptors on hepatocytes and engulfed by phagocytosis.
Remnants of VLDLs and Chylomicrons
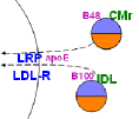
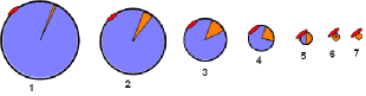
The illustration at the left is a section of the main diagram. The curved line represents the liver -- or an hepatocyte -- and the blue abbreviations are transporters embedded in its membrane. Both remnants will be taken into the hepatocyte by endocytosis as indicated by the dashed arrows.
The Space of Disse

The illustration at the right shows a large, porous capillary in the liver. The small particles are remnants. The line at the left in the illustration represents the hepatocyte membrane. The small circle at the top left represents a chylomicron remnant (CMR) and the one just below it is the remnant of VLDL called intermediate density lipoprotein (IDL). Notice the apoB48 and B100 apoproteins on the appropriate remnant. Also notice that the core of each contains approximately equal amounts of cholesterol ester (orange) and triglyceride (lavender).
Lipoprotein remnants are removed from circulation by the liver in three stages: entrance, sequestration and uptake.
Entrance
The capillaries in the liver are much larger in diameter than typical capillaries. Such vessels are called sinusoids. Additionally, there is a significant amount of space surrounding the sinusoids called the space of Disse. Small lipoproteins can enter and leave through the numerous openings (fenestrae) in the sinusoids.
Sequestration
The hepatocyte cell membrane has numerous large, branched molecules called heparin sulfate proteoglycans (HSPGs) (not shown) that extend into the space of Disse. These serve as attachment sites for many different particles. When the lipoprotein remnants contact them they become bound and sequester (confined) against the hepatic cell membrane.
Uptake
There are two types of receptors located in the hepatocyte membrane.
- Lipoprotein-receptor related protein (LRP) requires apoE to facilitates the binding of the sequestered remnants to this receptor. This is illustrated by 'apoE' found between 'LRP' and each type of remnant. ApoE is secreted into the space of Disse by the hepatocyte. Also, apoE might still be on the remnants if not lost when shedding excess coat material.
- The low density lipoprotein receptor (LDL-R) does not require apoE; it interacts with the B100 on IDLs as suggested in the illustration.
Upon attaching to the receptors the entire assembly is engulfed and taken into (endocytosis) the hepatocyte. Internally the lipoproteins are disassembled and the receptors are recycled back to the cell surface.
HDLs begin as small particles, become disc-shaped and mature to spheres. The newer, smaller spheres are most dense. HDLs pick up free cholesterol from cells and convert it into cholesterol ester (the 'not free' form of cholesterol). The cholesterol is taken back to the liver where it is used for bile.
High Density Lipoproteins
Unlike a triglyceride-rich low density lipoprotein, a high density lipoprotein starts out as a very small, disorganized particle of no specific shape. It progresses through a series of changes becoming a disc-shaped particle then it becomes spherical. Unlike low density lipoproteins its job is to gather instead of lose; it picks up cholesterol from various cells throughout the body. Eventually it returns to the liver -- where it started -- and deposits its cholesterol load there.
HDL Subfractions
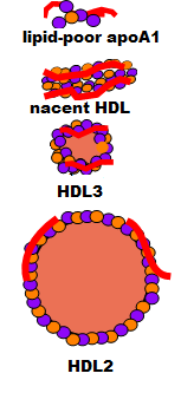
The illustration at the right shows how an HDL begins as an apoprotein A1 (red) plus a few molecules of phospholipid (purple) and free cholesterol (orange) secreted into the space of Disse. It has no particular organization; it is called lipid-poor apoA1. It accumulates additional phospholipid and free cholesterol molecules as it circulates and becomes a bilayered disc called nascent HDL. As it continues to circulate it picks up additional free cholesterol and phospholipids but now the free cholesterol is converted into cholesterol ester that is stored between the two layers and this converts it to a small sphere called HDL3. This process continues until the sphere grows, becoming less dense, and is called HDL2.
Lipid-poor ApoA1 to Nascent HDL
The illustration at the left is extracted from the main diagram to focus on how an HDL lipoprotein matures. The curved line at the top is part of an hepatocyte membrane. The liver secretes apoprotein A1, accompanied by a few cholesterol and phospholipid molecules, producing a small disorganized particle is called lipid-poor apoA1.
This particles leaves the space of Disse and enters the circulatory system. It is extremely small and readily passes through capillary walls coming into direct contact with cells throughout the body. Cells with excess cholesterol (small circle with C) have ATP-binding cassette transporter (ABCA-1) on their surface. This protein transports free cholesterol to the outer leaflet of the cell membrane. ApoA1 of the lipid-poor particles binds with ABCA-1 resulting in the transferred of free cholesterol and phospholipid to the particle.

As phospholipid and free cholesterol are added to the particle, these molecules spontaneously arrange themselves with the hydrophobic (water-hating) sections facing each other and their hydrophilic (water-loving) sections facing outward. The arrangement is like a typical double-leaflet cell membrane as shown to the right. The edge of this structure is encircled by the apoprotein A1 molecule that stabilizes the structure known as nascent HDL.
The plasma enzyme lecithin-cholesterol acyltransferase (LCAT) also binds to apoA1 but at a different site. Activated by this binding, the enzyme converts accumulating free cholesterol to cholesterol ester. It does this by removing a fatty acid from a phospholipid and transferring it to a neighboring free cholesterol -- the cholesterol is no longer 'free'. Being extremely hydrophobic, cholesterol ester accumulates between the two layers of the disc. This converts nascent HDL to a small sphere called HDL3. The conformation of apoA1 is altered when the disc becomes spherical causing its detachment from the ABCA-1.
HDL3
Cholesterol rich cells have scavenger receptor B-1 (SR-B1) transporters in addition to ABCA-1. Both function is essentially the same manner. As HDL particles contact these cells more cholesterol continues to accumulate and LCAT facilitates enlargement of the core.
To accommodate the expanding core, additional phospholipid and free cholesterol is also added to its coat by phospholipid transfer protein (PLTP). This protein accumulates sections of lipoprotein coat shed from other lipoproteins as they decrease in size. Included in these sections are also a variety of other apoproteins including additional apoA1s.
Reverse Cholesterol Transport
This ability of HDL to pick up cholesterol also applies to its encounter with cholesterol-rich foam cells. These cells reside just beneath the lining of inflamed arteries. SR-B1 transports cholesterol from intracellular droplets (circles with C) to the membrane. Additionally, foam cells have ABCG-1 transporters that do the same. HDL particles encountering these surfaces pick up more cholesterol and continue to enlarge. At some point the surface apoA1 becomes altered such that it no longer binds LCAT and no further enlargement can occur.
The density of the enlarging HDL eventually decreases to a point where it is called an HDL2. The '2' simply implies it is less dense than the '3' HDL. HDL1 particles are possible but they are rare in humans.
HDLs eventually pass through the liver sinusoids and enter the space of Disse. Here they encounter heparan sulfate proteoglycans (HSPG) bound to the hepatocyte membrane. These large, branched molecules have hepatic lipase (HL) bound to them. It appears that one of two things can happen when HDL and HL bind:
- The HL and nearby SR-B1 molecules work together so that the cholesterol ester in the core is selectively transferred into the hepatocyte leaving small particles resembling lipid-poor apoA1 in the space of Disse.
- The entire assemble of HDL/HL/SR-B1 is taken into the cell by endocytosis and disassembled with the apoA1 being resecreted and the SR-B1 recycled to the surface.
Collisions occur between lipoproteins in circulation. Core contents are exchanged along their concentration gradients with some particles enlarging and others shrinking. Of concern is the production of HDLs that are so small they can be lost via excretion by the kidneys.
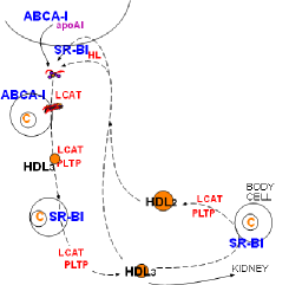
Lipoprotein Remodeling

At the lower left corner of the diagram is a small, cholesterol ester-rich HDL3. The previous tutorial described its conversion to a larger sphere after picking up free cholesterol from a cholesterol-rich body cell. The enlargement leading to HDL2 involves SR-B1, LCAT and PLTP .
'Good Cholesterol'
Included in this diagram is a large foam cell that has numerous cholesterol droplets. Foam cells form when macrophages become trapped beneath the endothelial lining of arteries and accumulate excessive cholesterol. If the droplets become too abundant an atherosclerotic plaque will develop. HDLs get their 'good cholesterol' label because they can remove free cholesterol from these cells just as they do from other cells.
In addition to the common SR-B1 transporter, foam cells have another transporter called ATP-binding cassette transporter G-1 (ABCG-1). This transporter functions in a manner similar to that of ABCA-1. It places rafts of free cholesterol in the foam cell's outer leaflet. LCAT facilitate the conversion of the new free cholesterol into cholesterol ester and PLTP adds additional coat material as sphere continues to enlarge.
Lipoprotein Interactions
There are frequent collisions (indicated by curved arrows coming together) among the various lipoproteins while circulating. There is evidence that a plasma protein called cholesterol ester transfer protein (CETP) binds lipoproteins and forms a hydrophobic channel between their cores. This allows diffusion of core lipids -- triglyceride and cholesterol ester -- to be exchanged between triglyceride-rich and cholesterol ester-rich lipoproteins.
As an example, the diagram shows what would result if an IDL and an HDL were involved in such a collision. Clearly the triglyceride (lavender) gradient favors diffusion from the IDL into the HDL and the cholesterol ester gradient favors diffusion in the opposite direction. After separation the IDL is shown having less triglyceride and more cholesterol ester while the HDL now has some triglyceride in its core.
In addition to the change in core composition of each particle there is also a size change. Because a triglyceride molecule occupies more space than a cholesterol ester molecule the HDL has enlarged and the IDL has shrunk. Note this change in the diagram.
Hepatic lipase (HL) is a plasma enzyme that hydrolyzes triglycerides into free fatty acids and glycerides. It also has phospholipid activity and can destroy lipoprotein coats. In this diagram it is shown changing a triglyceride-rich HDL to a smaller HDL3 with no triglyceride left.
A big concern is that an increase in CETP activity will create more HDLs with triglyceride in their core. Since triglyceride will be hydrolyzed by hepatic lipase the particle will shrink. The more triglyceride, the more it will shrink. Then, the smaller the particle the more likely it will be excreted by the kidneys. This means a loss in HDL -- the 'good cholesterol'.
LDLs are formed from IDLs due to the catalytic activity of hepatic lipase. This enzyme is bound to hepatocytes in an inactive form. HL is detached by HDLs and transferred to triglyceride-rich lipoproteins where it actively hydrolyze triglyceride and shrinks the lipoprotein. 'Reverse cholesterol transport' is when HDLs return cholesterol to the liver.
Low Density Lipoprotein
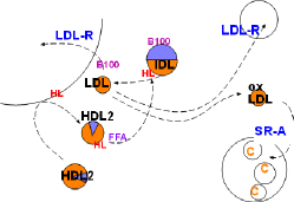
Low density lipoproteins (LDLs) are formed from intermediate density lipoproteins (IDLs) shown in the top center of the diagram. This conversion is due to the catalytic activity of hepatic lipase (HL, red) inscribed on the arrow. This enzyme hydrolyzes any triglyceride in the IDL and also removes excess phospholipids from the IDL coat as it shrinks. (Note relative sizes of the IDL and LDL).
Hepatic Lipase
Hepatic lipase resides on HSPGs (not shown) on hepatocyte membranes and is inactive. It must be dislodged from the HSPGs, transported into circulation and activated at the appropriate time. The catalytic activities of hepatic lipase would destroy cell membranes if it circulated in an active form. However, it is readily accessible, though inactive, while bound to hepatocytes (HL on curved line (hepatocyte) at upper left ).
Hepatic lipase is most effectively dislodged by the larger types of HDL (HDL2) This is shown by the HDL2 from the lower left contacting the hepatic lipase (HL) attached to the hepatocyte (upper left). From there HDL leaves, with the HL attached, to re-enter the general circulation. The HL is still inactive.
Hepatic lipase can be transferred to other lipoproteins under the right conditions. It does not circulate freely in the plasma. There are several possible explanations as to what causes this transfer. One possibility is the increase of plasma free fatty acids after a meal.
When hepatic lipase is transferred to the recipient lipoprotein it becomes active hydrolyzing triglyceride and reducing the phospholipid in the coat. The lipoprotein decreases in size and becomes triglyceride-poor -- and therefore cholesterol ester-rich. The diagram represents these events showing an IDL with a core half triglyceride (lavender) and half phospholipid (orange)being converted to a smaller cholesterol ester-rich low density lipoprotein (LDL).
Cholesterol Transport
Low density lipoprotein is small enough to penetrate capillary walls and deliver cholesterol esters to those cells displaying low density lipoprotein receptors (LDL-R). This receptor binds to apoprotein B100 on the particles resulting in phagocytosis.
The liver displays abundant LDL-R receptors and accounts for most LDL uptake. Body cells that produce steroids also have a constant need for cholesterol as shown in the upper right of the diagram.
Unfortunately, low density lipoproteins also enter inflamed areas of arteries. There macrophages secrete numerous chemicals involved in the inflammatory response and the LDLs become oxidized. Scavenger receptor A (SR-A), abundant on active macrophages, specifically binds with oxidized LDLs (oxLDL) and phagocytize them. This increase in cholesterol within macrophages converts them to foam cells. The foamy appearance is due to the accumulation of lipid droplets in their cytoplasm (lower right).
Summary
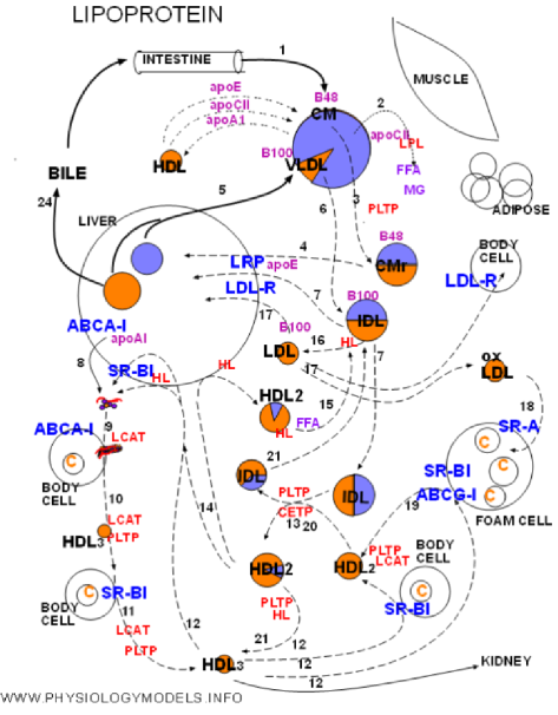
The physiology of lipoproteins will be traced from the entrance of chylomicrons through the return of cholesterol to the liver.
The numbers on the arrows will allow you to match the diagram with the following narrative.
1. Chylomicrons bring in dietary triglyceride and cholesterol ester from the intestine. These are the largest of the lipoproteins. They have apoprotein 48 on entry but will pick up apoE and apoCII from HDLs and donate apoA1 to them. Triglyceride is by far the most abundant core content.
2. During circulation chylomicrons will encounter lipoprotein lipase (LPL). The inactive enzyme is secreted by muscle and adipose tissue under hormonal control. This enzyme will be activated when bound to apoCII on the chylomicron resulting in rapid hydrolysis of core triglyceride. The products are free fatty acid (FFA) and monoglyceride (MG).
3. Reduction of the core volume reduces the size of the chylomicron forming a remnant. The excess coat material is transferred to phospholipid transfer protein (PLTP).
4. Chylomicron remnants become bound to lipoprotein receptor-related protein (LRP). Apoprotein E, secreted by the liver, is required for binding and the result is phagocytosis.
5. The liver secretes very low density lipoproteins (VLDLs) with approximately the same proportion of core contents as chylomicrons. The triglyceride and cholesterol ester come from intracellular storage sites. Apoprotein B100 is part of the coat.
6. The VLDL, secreted with apoCII as part of its coat, encounters LPL and activates it to hydrolyze its triglyceride. As with chylomicrons there is a reduction in size and loss of excess coat material. The remnant is called intermediate density lipoprotein (IDL).
7. IDL is phagocytized by the liver after, in association with apoE, binding to LRP. It also binds with low density lipoprotein receptor (LDL-R) that recognizes apoB100.
8. High density lipoproteins begin as apoprotein A1 secreted by the liver. The protein ATP-binding cassette A-1 (ABCA-1) transferred excess cholesterol from intracellular storage areas to the hepatocyte membrane. ApoA1 binds with this protein and picks up a few molecules of free cholesterol and phospholipid from the hepatocyte outer leaflet. The disorganized particle called lipid-poor A1 is released into circulation.
9. While circulating, lipid-poor A1 contacts body cells with ABCA-1. Apoprotein A1 of the disorganized particle binds with ABCA-1 resulting in sections of the cells outer leaflet being transferred to the particle. The additional phospholipid and free cholesterol molecules spontaneously form a bi-layered disc-shaped particle called nascent HDL. LCAT on the nacent HDL converts free cholesterol into cholesterol ester that forms a core.
10&11. The circulating small HDL3 accumulates additional cholesterol transferred to the membrane by scavenger receptor B-1 (SR-B1). Contact results in diffusion of cholesterol to HDL. LCAT continues to increase the core. PLTP supplies coat material.
12. HDL3 can follow four paths:
- excretion via kidneys if still small,
- continue accumulating cholesterol from body cells and enlarging,
- pick up cholesterol from foam cells and enlarging,
- transport accumulated cholesterol to the liver
13. Lipoprotein remodeling is exchange of triglyceride and cholesterol ester between lipoproteins along concentration gradients. Particles gaining triglyceride enlarge and those gaining cholesterol ester shrink. CETP and PLTP facilitate this process.
14. Larger HDL2 particles return to the liver where they either deliver cholesterol ester by selective uptake mediated by SR-B1 or are endocytosed after complexing with hepatic lipase and SR-B1 Either way apoA1 is recycled. Alternatively they may liberate hepatic lipase.
15. Hepatic lipase on HDLs is inactive until transferred to triglyceride-rich B100 particles. This occurs after meals (FFA) and the enzyme becomes active.
16. Triglycerides in IDLs are hydrolyzed by hepatic lipase forming LDLs.
17. LDLs can follow three paths:
- phagocytosis by the liver via LDL-R
- phagocytosis by body cells via LDL-R
- oxidation in the presence of foam cells.
18. Oxidized LDL is phagocytized by foam cells via SR-A thus facilitating atherosclerosis.
19. Foam cells transport cholesterol to their surface by SR-B1 and ABCG-1 to be picked up by HDL particles.
20. Large lipid exchanges during remodeling would result in triglyceride-rich HDLs and cholesterol ester-rich IDLs.
21. Triglyceride-rich HDLs and cholesterol ester-rich IDLs can be hydrolyzed by hepatic lipase resulting in smaller than usual particles.